OLÁ PESSOAL DO LABORATÓRIO,
Neste texto vamos mostrar os principais pontos sobre a Doença de Gaucher.
A Doença de Gaucher é uma patologia de ordem genética, caracterizada por uma acumulação de “glicocerebrosídeos” (um espécie de substância de caráter gorduroso) que ocorre em células reticulo-endoteliais.
Só relembrando, essas células são as que compõem o sistema reticulo-endotelial, ou sistema mononuclear fagocitário, que tem características reticulares e endoteliais, e tem uma capacidade fagocitaria.
Elas estão situadas em diversos locais do organismo, como por exemplo, os macrófagos que encontramos na medula óssea, que é o que vamos falar aqui, correlacionando ao assunto.
Então pessoal, o acúmulo dessa substância, ocorre devido uma deficiência da enzima glicocerebrosidase.
E com a atividade reduzida dessa enzima, o glicocerebrosídeo, que é a substância que deveria ser “quebrada pela enzima” não é digerido dentro do lisossomo, e se acumula progressivamente nos macrófagos.
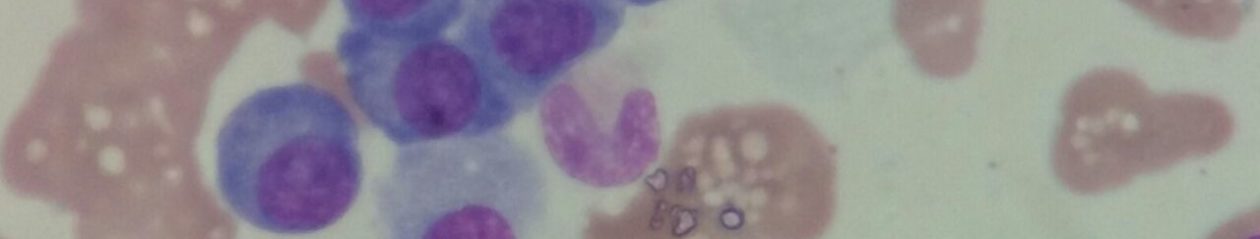
Estes, os macrófagos – que são as células reticulo-endoteliais – aumentam de tamanho e passam a se chamar “Células de Gaucher”.
As células de Gaucher, por sua vez, se acumulam nos tecidos, e isso ocorre principalmente no fígado e no baço, e estes órgãos, consequentemente também aumentam de tamanho, o que chamamos de hepatomegalia e esplenomegalia, respectivamente.
Essas células, as de Gaucher, também se encontram na medula óssea (internamente, na porção óssea), causando um enfraquecimento do osso, o que pode levar a fraturas com mais facilidade que ossos normais.
A CLASSIFICAÇÃO DA DOENÇA
Os tipos da doença de Gaucher: são clinicamente diferenciados com base na ausência (tipo 1) ou na presença de manifestações neurológicas (tipos 2 e 3), e a doença depende do depósito da substância nos tecidos, que podem ocorrer em diferentes órgãos.
Sumariamente, temos a divisão da seguinte forma:
· tipo I: ausência de sinais de envolvimento do sistema nervoso central
· tipo II: tipo agudo neuropático infantil;
· tipo III: tipo subagudo neuropático
O tipo III ainda tem algumas subdivisões que não vamos abordar aqui, mas que se vocês quiserem aprofundar esse estudo, podem pesquisar nas Referências bibliográficas que iremos deixar no fim do texto.
·
A doença de Gaucher tem uma gravidade muito variável;
A doença do tipo 1 pode ser assintomática e ser descoberta acidentalmente em levantamentos genéticos populacionais. A fadiga é uma queixa comum, geralmente não relacionada à anemia, mas também bastante comum em pacientes anêmicos, e pode resultar de citocinas elevadas.
Em pacientes sintomáticos, o baço é tipicamente aumentado.
A hepatomegalia é comum, geralmente assintomática, mas ocorre em grau menor que a esplenomegalia.
A linfadenopatia é descrita na literatura, mas não é um achado marcante.
Eventos hemorrágicos como epistaxe, hematomas e hemorragia com procedimentos cirúrgicos ou odontológicos e sangramento durante o trabalho de parto são sintomas comuns.
Essas manifestações, geralmente, estão relacionadas à trombocitopenia que é uma consequência do hiperesplenismo ou substituição de medula óssea pelas células de Gaucher, mas a disfunção plaquetária e a diminuição dos níveis dos fatores de coagulação também foram descritas e, portanto, devem ser avaliadas antes do procedimento cirúrgico ou antes do parto. As deficiências do fator de coagulação podem ocorrer devido à doença hepática ou à coagulopatia por consumo.
Níveis reduzidos de hemoglobina podem ocorrer devido ao hiperesplenismo e à substituição da medula pelas células de Gaucher, mas também observam-se outras causas. Incluem deficiência de ferro, deficiência de vitamina B12 e hemólise autoimune. O depósito de glicopeptídeos em órgãos pode formar neoplasias primárias benignas denominadas de Gaucheromas, podendo imitar um processo maligno.
Doença pulmonar grave com cianose e hipoxemia ocorre em alguns pacientes com comprometimento hepático avançado, e costuma ser uma consequência da síndrome hepatopulmonar com ou sem infiltração pulmonar pelas células de Gaucher, que pode ocluir os vasos pulmonares.
O acometimento ósseo é a principal causa de morbidade em pacientes sintomáticos e pode ocorrer em qualquer osso longo. Observam-se áreas de desmineralização e infarto ósseo e alargamento assintomático do fêmur distal com importante dor óssea, podendo cursar com crises de dor óssea que podem ter como desfecho necrose avascular óssea.
Em resumo, em relação à sintomatologia podemos observar os principais pontos:
Ao exame clínico o paciente pode apresentar hepatoesplenomegalia, anemia, trombocitopenia e deterioração esquelética.
Além disso são observados outros achados comuns: palidez, cansaço, fraqueza, em decorrência da anemia,
E dores nas pernas e ossos, com propensão a fraturas, inclusive com pancadas leves, devido a fragilidade óssea, também já comentada.
EXAMES COMPLEMENTARES
O hemograma em pacientes com doença de Gaucher pode ser normal ou refletir os efeitos do hiperesplenismo.
Uma anemia normocítica e normocrômica está frequentemente presente, mas os níveis de hemoglobina raramente caem abaixo de 8g/dl.
Uma modesta reticulocitose está presente em pacientes anêmicos.
A contagem de leucócitos pode ser reduzida para até 1.000 células/mm3, mas graus mais leves de leucopenia são mais comuns.
A contagem diferencial é normal, mas os pacientes esplenectomizados tendem a apresentar linfocitose.
A trombocitopenia pode ser bastante grave.
Anisocitose e poiquilocitose também ocorrem em pacientes esplenectomizados, com muitas células em alvo e corpos de Howell-Jolly.
Durante as crises ósseas, ocorre leucocitose, trombocitose e aumento da velocidade de hemossedimentação (VHS) são observados.
Aumento de marcadores inflamatórios podem ocorrer, independentemente da gravidade da doença, assim como níveis elevados de fibrinogênio, proteína C-reativa e aumento da adesão e agregação de hemácias.
Anormalidades no fator de coagulação podem ser induzidas por macrófagos ativados ou podem ser encontradas quando há comprometimento hepático.
As enzimas hepáticas são discretamente elevadas. A hiperferritinemia, nesses pacientes, pode ser significativa.
A tendência ao sangramento também pode resultar da agregação ou adesão defeituosa das plaquetas, e a função plaquetária deve ser testada antes dos procedimentos cirúrgicos e odontológicos e do parto.
Os testes de função renal, geralmente são normais.
Muitos pacientes apresentam gamopatias policlonais, e gamopatias monoclonais são encontradas em graus que variam de 1 a 20% dos pacientes, particularmente em idosos.
Níveis aumentados de autoanticorpos foram relatados e podem coincidir com doenças autoimunes, como a tireoidite de Hashimoto, artrite reumatoide ou anemia hemolítica imunológica.
A avaliação de medula, baço e fígado pode demonstrar a presença de células de Gaucher.
Elas podem também ser encontradas em outros tecidos, e a presença é o achado característico da doença.
DIAGNÓSTICO DIFERENCIAL
O diagnóstico da doença de Gaucher deve ser considerado em qualquer paciente que apresente esplenomegalia sem outra causa, trombocitopenia, frequentes hemorragias nasais, anemia, dor óssea aguda ou crônica, crianças com baixa estatura para a idade; e necrose avascular não traumática de uma articulação grande em qualquer idade, especialmente se estiver associada a qualquer uma das características supracitadas.
Um diagnóstico definitivo requer uma atividade enzimática reduzida da ß-glicocerebrosidase em leucócitos, fibroblastos cultivados ou amniócitos obtidos durante o diagnóstico pré-natal.
A mensuração dos níveis de glicocerebrosidase é complementada por análise genética procurando as mutações associadas à doença.
A aspiração de medula óssea como meio de diagnóstico só é indicada quando outras doenças hematológicas devem ser consideradas. Células de Gaucher são escassas e podem não ser encontradas na biópsia de medula óssea.
E aí? Gostou? Então compartilha com um colega que precisa conhecer esse assunto.
É isso aí pessoal,
“Tamo junto” nessa jornada hematológica.
Um abraço, até a próxima.
Bibliografia
1-Zimran A, ElsteinD. Gaucher?s Disease and related lisossomal storage Diseases in WilliamsHematology 2016.
2-Zimran A. How I treat Gaucher disease. Blood 2011; 118:1463.
3- http://www.gaucher.org.uk/enews.php?id=303 acesso 18/07/2019.
4- Martins AM, Valadares ER, PortaG, et al. Recommendations on diagnosis,treatment, and monitoring for Gaucher disease. J Pediatr 2009; 155:S10.